正在加载图片...
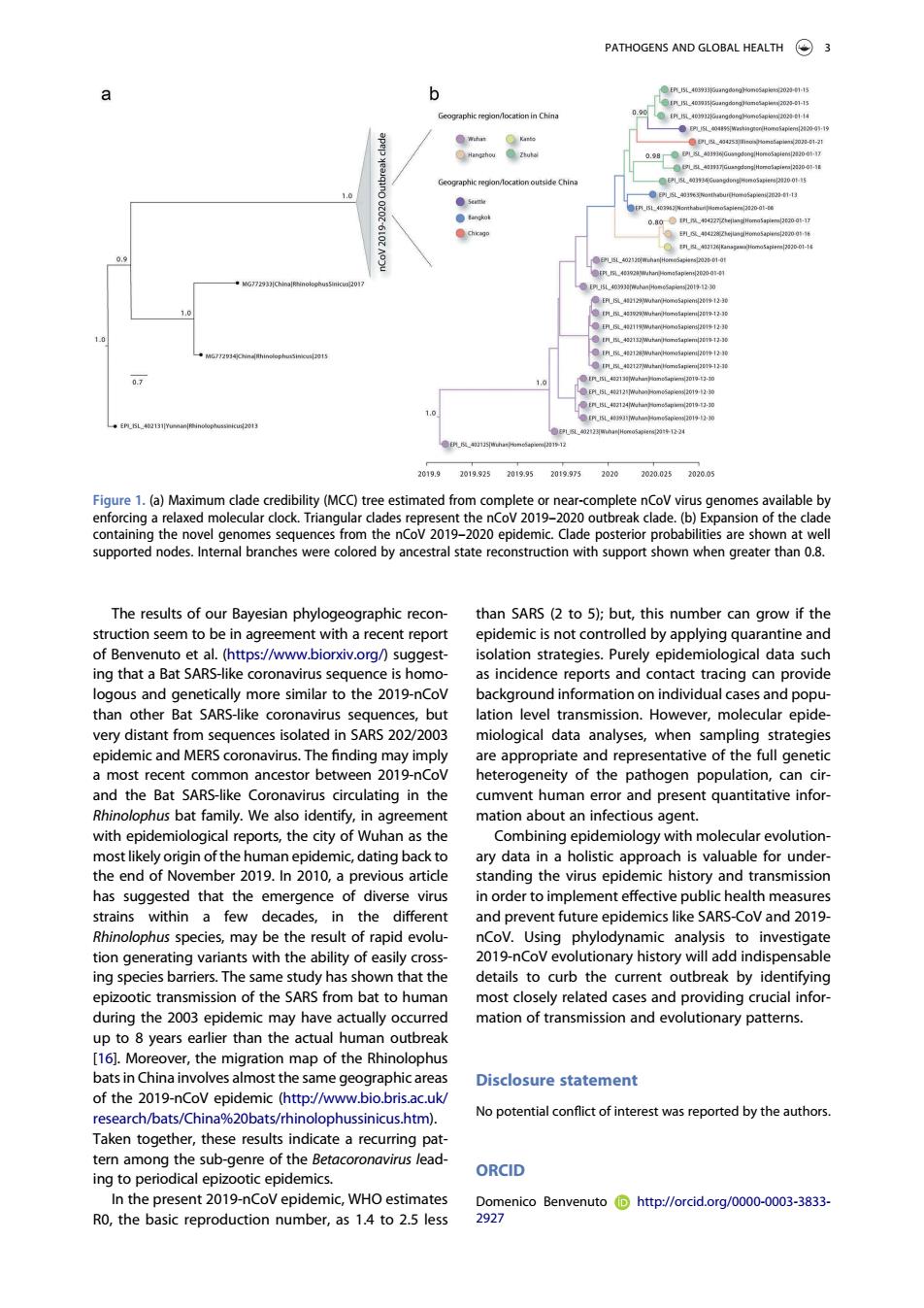
PATHOGENS AND GLOBAL HEALTH 3 人 5oquenceshomhenCo/2019-2z00e9pdemLa39eOp0ekp6blfnieonc Aa The results of our Bavesian phylogeographic recon- than SARS(2 to 5):but,this number can grow if the struction seem to be in agreement with a recent report epidemic is not controlled by applying quarantine and to et al.(https://www.biorxiv.org/)suggest isolation strategies.Purely epidemiological data such s sequ as inc ence reports and than other Bat SARS-like quences.but very distant from sequences isolated in SARS 202/2003 miological data analyses,when sampling strategies epidemic and MERS coronavirus.The finding may imply are appropriate and representative of the full genetic 201 ity of the pat ating in th mvent huma pre hus hat fa tio Combining epidemioloay with molecular evolution most human epidemic,dating backto ary data in a holistic approach is valuable for under the end of N vember 2019.n2010,a previous article standing the virus epid emic history and transmission has sugg emergence n me 201 may he the nCoV.Using e to tion generatina variants with the ability of easily cros 201no evolutionary historywil add indispensable ing species barriers.The same study has shown that the details to curb the current outbreak by identifying epizootic transmission of the SARS from bat to huma ost closely related cases and providing crucial infor ng the 200 mation of transmission and evolutionary patterns ,h3 161.Mo er the ration man of the Rhi bats in China involves almost the same geographicareas Disclosure statement of the 2019-nCoV epidemic (http://www.bio.bris.ac.uk/ No potential conflict of interest was reported by the authors arcn/bat a%20b phussinicus.htm ese res ORCID In the present 2019-nCoV epidemic.WHO estimates RO,the basic reproduction number,as 1.4 to 2.5 less g7ntoBememuio自hmpr/orcdog0o00o3383a The results of our Bayesian phylogeographic reconstruction seem to be in agreement with a recent report of Benvenuto et al. (https://www.biorxiv.org/) suggesting that a Bat SARS-like coronavirus sequence is homologous and genetically more similar to the 2019-nCoV than other Bat SARS-like coronavirus sequences, but very distant from sequences isolated in SARS 202/2003 epidemic and MERS coronavirus. The finding may imply a most recent common ancestor between 2019-nCoV and the Bat SARS-like Coronavirus circulating in the Rhinolophus bat family. We also identify, in agreement with epidemiological reports, the city of Wuhan as the most likely origin of the human epidemic, dating back to the end of November 2019. In 2010, a previous article has suggested that the emergence of diverse virus strains within a few decades, in the different Rhinolophus species, may be the result of rapid evolution generating variants with the ability of easily crossing species barriers. The same study has shown that the epizootic transmission of the SARS from bat to human during the 2003 epidemic may have actually occurred up to 8 years earlier than the actual human outbreak [16]. Moreover, the migration map of the Rhinolophus bats in China involves almost the same geographic areas of the 2019-nCoV epidemic (http://www.bio.bris.ac.uk/ research/bats/China%20bats/rhinolophussinicus.htm). Taken together, these results indicate a recurring pattern among the sub-genre of the Betacoronavirus leading to periodical epizootic epidemics. In the present 2019-nCoV epidemic, WHO estimates R0, the basic reproduction number, as 1.4 to 2.5 less than SARS (2 to 5); but, this number can grow if the epidemic is not controlled by applying quarantine and isolation strategies. Purely epidemiological data such as incidence reports and contact tracing can provide background information on individual cases and population level transmission. However, molecular epidemiological data analyses, when sampling strategies are appropriate and representative of the full genetic heterogeneity of the pathogen population, can circumvent human error and present quantitative information about an infectious agent. Combining epidemiology with molecular evolutionary data in a holistic approach is valuable for understanding the virus epidemic history and transmission in order to implement effective public health measures and prevent future epidemics like SARS-CoV and 2019- nCoV. Using phylodynamic analysis to investigate 2019-nCoV evolutionary history will add indispensable details to curb the current outbreak by identifying most closely related cases and providing crucial information of transmission and evolutionary patterns. Disclosure statement No potential conflict of interest was reported by the authors. ORCID Domenico Benvenuto http://orcid.org/0000-0003-3833- 2927 Figure 1. (a) Maximum clade credibility (MCC) tree estimated from complete or near-complete nCoV virus genomes available by enforcing a relaxed molecular clock. Triangular clades represent the nCoV 2019–2020 outbreak clade. (b) Expansion of the clade containing the novel genomes sequences from the nCoV 2019–2020 epidemic. Clade posterior probabilities are shown at well supported nodes. Internal branches were colored by ancestral state reconstruction with support shown when greater than 0.8. PATHOGENS AND GLOBAL HEALTH 3