正在加载图片...
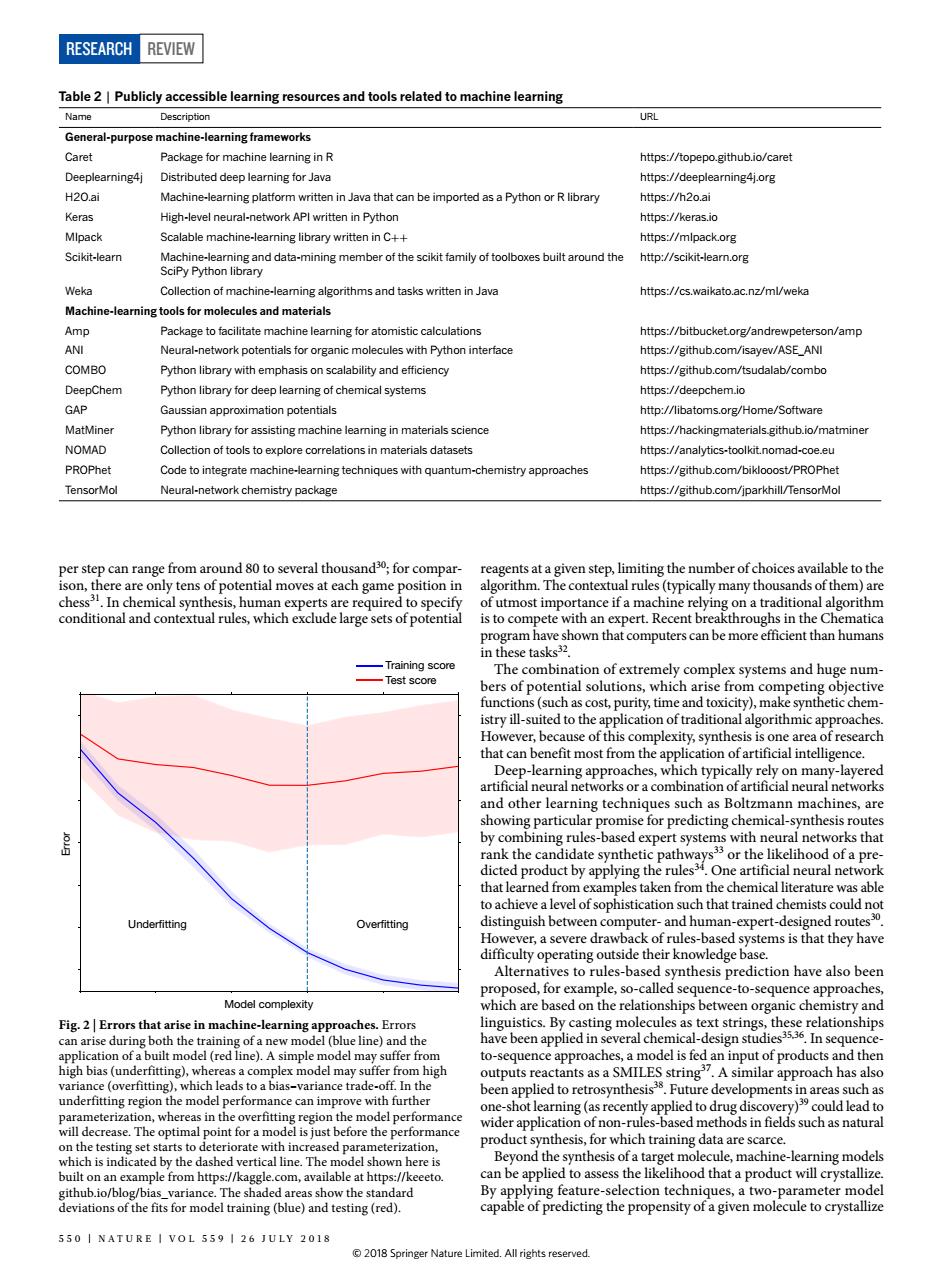
RESEARCH REVIEW Table2 Publicly accessible learning resources and tools related to machine learning Name URL General-purpose machine-learningframeworks age for mac ning in F https://topepo.github.io/caret arning4j.org Java that can be ://h2oai Scalable machine-eaming lbary wrtten inC httns://mipackore Scikit-leamn http//scikit-learnore Weka aming algorithms and tasks written in Java https://cs.waikato.ac.nz/ml/weka arning tools for molecules and materials Amp Package to facilitate machine learning for atomistic calculations https://bitbucketorg/andrewpeterson/amp ote Tor organic molecules with Python interfac GAP MatMiner Python library for assisting machine leaming in materials science https://hackingmaterials.github.io/matmine NOMAD Collection of tools to explore correlations in materials datasets https://analytics-toolkitnomad-c PROPhet Code to integrate machine-learning techniques with quantum-chemistry approaches nttps://github.com/biklooost/PROPhe TensorMo Neural-network chemistry package https://github.com/jparkhill/TensorMo ks that Underfitting approach Fig.2E 50 I NATURE I VOL 559 1 26 JULY 2018 2018RESEARCH Review Table 2 | Publicly accessible learning resources and tools related to machine learning Name Description URL General-purpose machine-learning frameworks Caret Package for machine learning in R https://topepo.github.io/caret Deeplearning4j Distributed deep learning for Java https://deeplearning4j.org H2O.ai Machine-learning platform written in Java that can be imported as a Python or R library https://h2o.ai Keras High-level neural-network API written in Python https://keras.io Mlpack Scalable machine-learning library written in C++ https://mlpack.org Scikit-learn Machine-learning and data-mining member of the scikit family of toolboxes built around the SciPy Python library http://scikit-learn.org Weka Collection of machine-learning algorithms and tasks written in Java https://cs.waikato.ac.nz/ml/weka Machine-learning tools for molecules and materials Amp Package to facilitate machine learning for atomistic calculations https://bitbucket.org/andrewpeterson/amp ANI Neural-network potentials for organic molecules with Python interface https://github.com/isayev/ASE_ANI COMBO Python library with emphasis on scalability and efciency https://github.com/tsudalab/combo DeepChem Python library for deep learning of chemical systems https://deepchem.io GAP Gaussian approximation potentials http://libatoms.org/Home/Software MatMiner Python library for assisting machine learning in materials science https://hackingmaterials.github.io/matminer NOMAD Collection of tools to explore correlations in materials datasets https://analytics-toolkit.nomad-coe.eu PROPhet Code to integrate machine-learning techniques with quantum-chemistry approaches https://github.com/biklooost/PROPhet TensorMol Neural-network chemistry package https://github.com/jparkhill/TensorMol Training score Test score Underfitting Overfitting Model complexity Error Fig. 2 | Errors that arise in machine-learning approaches. Errors can arise during both the training of a new model (blue line) and the application of a built model (red line). A simple model may suffer from high bias (underfitting), whereas a complex model may suffer from high variance (overfitting), which leads to a bias–variance trade-off. In the underfitting region the model performance can improve with further parameterization, whereas in the overfitting region the model performance will decrease. The optimal point for a model is just before the performance on the testing set starts to deteriorate with increased parameterization, which is indicated by the dashed vertical line. The model shown here is built on an example from https://kaggle.com, available at https://keeeto. github.io/blog/bias_variance. The shaded areas show the standard deviations of the fits for model training (blue) and testing (red). reagents at a given step, limiting the number of choices available to the algorithm. The contextual rules (typically many thousands of them) are of utmost importance if a machine relying on a traditional algorithm is to compete with an expert. Recent breakthroughs in the Chematica program have shown that computers can be more efficient than humans in these tasks32. The combination of extremely complex systems and huge numbers of potential solutions, which arise from competing objective functions (such as cost, purity, time and toxicity), make synthetic chemistry ill-suited to the application of traditional algorithmic approaches. However, because of this complexity, synthesis is one area of research that can benefit most from the application of artificial intelligence. Deep-learning approaches, which typically rely on many-layered artificial neural networks or a combination of artificial neural networks and other learning techniques such as Boltzmann machines, are showing particular promise for predicting chemical-synthesis routes by combining rules-based expert systems with neural networks that rank the candidate synthetic pathways33 or the likelihood of a predicted product by applying the rules34. One artificial neural network that learned from examples taken from the chemical literature was able to achieve a level of sophistication such that trained chemists could not distinguish between computer- and human-expert-designed routes30. However, a severe drawback of rules-based systems is that they have difficulty operating outside their knowledge base. Alternatives to rules-based synthesis prediction have also been proposed, for example, so-called sequence-to-sequence approaches, which are based on the relationships between organic chemistry and linguistics. By casting molecules as text strings, these relationships have been applied in several chemical-design studies35,36. In sequenceto-sequence approaches, a model is fed an input of products and then outputs reactants as a SMILES string37. A similar approach has also been applied to retrosynthesis38. Future developments in areas such as one-shot learning (as recently applied to drug discovery)39 could lead to wider application of non-rules-based methods in fields such as natural product synthesis, for which training data are scarce. Beyond the synthesis of a target molecule, machine-learning models can be applied to assess the likelihood that a product will crystallize. By applying feature-selection techniques, a two-parameter model capable of predicting the propensity of a given molecule to crystallize per step can range from around 80 to several thousand30; for comparison, there are only tens of potential moves at each game position in chess31. In chemical synthesis, human experts are required to specify conditional and contextual rules, which exclude large sets of potential 550 | NATUR E | V OL 559 | 26 J U LY 2018 © 2018 Springer Nature Limited. All rights reserved