正在加载图片...
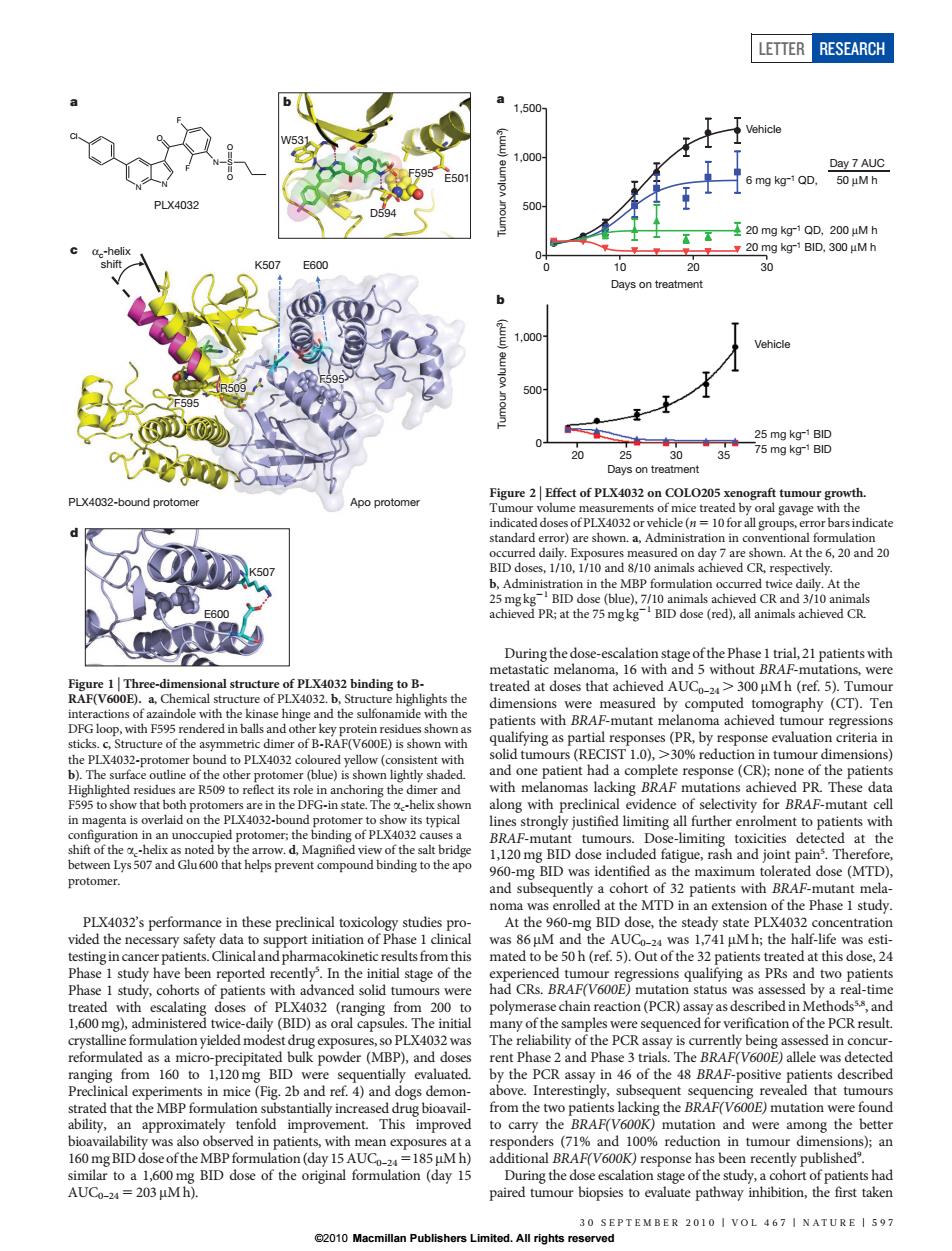
LETTER RESEARCH 20 Days on treatmen 500 20 PLX4032-bound protome Figure 2 Elte t of PL ca vn.At the 6,20 and 20 BID do At the V600 a of the dimer of B-RAF(V600E)sshe to PL wit (blue)i at the 1 PLX4032's perfom nce in these precinical toxicology studies pro At the 960 centratio study ha so PLX4032 was er(MBP).d the PCR assay in 46 of the sitive approxima nse has been r 241PLX4032’s performance in these preclinical toxicology studies provided the necessary safety data to support initiation of Phase 1 clinical testingin cancer patients.Clinical and pharmacokinetic resultsfrom this Phase 1 study have been reported recently5 . In the initial stage of the Phase 1 study, cohorts of patients with advanced solid tumours were treated with escalating doses of PLX4032 (ranging from 200 to 1,600 mg), administered twice-daily (BID) as oral capsules. The initial crystalline formulation yielded modest drug exposures, so PLX4032 was reformulated as a micro-precipitated bulk powder (MBP), and doses ranging from 160 to 1,120 mg BID were sequentially evaluated. Preclinical experiments in mice (Fig. 2b and ref. 4) and dogs demonstrated that the MBP formulation substantially increased drug bioavailability, an approximately tenfold improvement. This improved bioavailability was also observed in patients, with mean exposures at a 160 mg BID dose of theMBPformulation (day 15 AUC0–24 5185 mM h) similar to a 1,600 mg BID dose of the original formulation (day 15 AUC0–24 5 203 mM h). During the dose-escalation stage of the Phase 1 trial, 21 patients with metastatic melanoma, 16 with and 5 without BRAF-mutations, were treated at doses that achieved AUC0–24 . 300 mM h (ref. 5). Tumour dimensions were measured by computed tomography (CT). Ten patients with BRAF-mutant melanoma achieved tumour regressions qualifying as partial responses (PR, by response evaluation criteria in solid tumours (RECIST 1.0), .30% reduction in tumour dimensions) and one patient had a complete response (CR); none of the patients with melanomas lacking BRAF mutations achieved PR. These data along with preclinical evidence of selectivity for BRAF-mutant cell lines strongly justified limiting all further enrolment to patients with BRAF-mutant tumours. Dose-limiting toxicities detected at the 1,120 mg BID dose included fatigue, rash and joint pain5 . Therefore, 960-mg BID was identified as the maximum tolerated dose (MTD), and subsequently a cohort of 32 patients with BRAF-mutant melanoma was enrolled at the MTD in an extension of the Phase 1 study. At the 960-mg BID dose, the steady state PLX4032 concentration was 86 mM and the AUC0–24 was 1,741 mM h; the half-life was estimated to be 50 h (ref. 5). Out of the 32 patients treated at this dose, 24 experienced tumour regressions qualifying as PRs and two patients had CRs. BRAF(V600E) mutation status was assessed by a real-time polymerase chain reaction (PCR) assay as described in Methods5,8, and many of the samples were sequenced for verification of the PCR result. The reliability of the PCR assay is currently being assessed in concurrent Phase 2 and Phase 3 trials. The BRAF(V600E) allele was detected by the PCR assay in 46 of the 48 BRAF-positive patients described above. Interestingly, subsequent sequencing revealed that tumours from the two patients lacking the BRAF(V600E) mutation were found to carry the BRAF(V600K) mutation and were among the better responders (71% and 100% reduction in tumour dimensions); an additional BRAF(V600K) response has been recently published9 . During the dose escalation stage of the study, a cohort of patients had paired tumour biopsies to evaluate pathway inhibition, the first taken PLX4032 a N N O F F N S O O Cl D594 E501 F595 W531 c PLX4032-bound protomer Apo protomer F595 αc-helix shift F595 R509 K507 E600 E600 K507 d b Figure 1 | Three-dimensional structure of PLX4032 binding to BRAF(V600E). a, Chemical structure of PLX4032. b, Structure highlights the interactions of azaindole with the kinase hinge and the sulfonamide with the DFG loop, with F595 rendered in balls and other key protein residues shown as sticks. c, Structure of the asymmetric dimer of B-RAF(V600E) is shown with the PLX4032-protomer bound to PLX4032 coloured yellow (consistent with b). The surface outline of the other protomer (blue) is shown lightly shaded. Highlighted residues are R509 to reflect its role in anchoring the dimer and F595 to show that both protomers are in the DFG-in state. The ac-helix shown in magenta is overlaid on the PLX4032-bound protomer to show its typical configuration in an unoccupied protomer; the binding of PLX4032 causes a shift of the ac-helix as noted by the arrow. d, Magnified view of the salt bridge between Lys 507 and Glu 600 that helps prevent compound binding to the apo protomer. a 0 10 20 30 0 500 1,000 1,500 Days on treatment Tumour volume (mm3) Vehicle 6 mg kg–1 QD, 20 mg kg–1 QD, 200 μM h 20 mg kg–1 BID, 300 μM h Day 7 AUC Vehicle 25 mg kg–1 BID 75 mg kg–1 BID b 50 μM h 20 30 0 500 1,000 Days on treatment Tumour volume (mm3) 25 35 Figure 2 | Effect of PLX4032 on COLO205 xenograft tumour growth. Tumour volume measurements of mice treated by oral gavage with the indicated doses of PLX4032 or vehicle (n 5 10 for all groups, error bars indicate standard error) are shown. a, Administration in conventional formulation occurred daily. Exposures measured on day 7 are shown. At the 6, 20 and 20 BID doses, 1/10, 1/10 and 8/10 animals achieved CR, respectively. b, Administration in the MBP formulation occurred twice daily. At the 25 mg kg21 BID dose (blue), 7/10 animals achieved CR and 3/10 animals achieved PR; at the 75 mg kg21 BID dose (red), all animals achieved CR. LETTER RESEARCH 30 SEPTEMBER 2010 | VOL 467 | NATURE | 597 ©2010 Macmillan Publishers Limited. All rights reserved