正在加载图片...
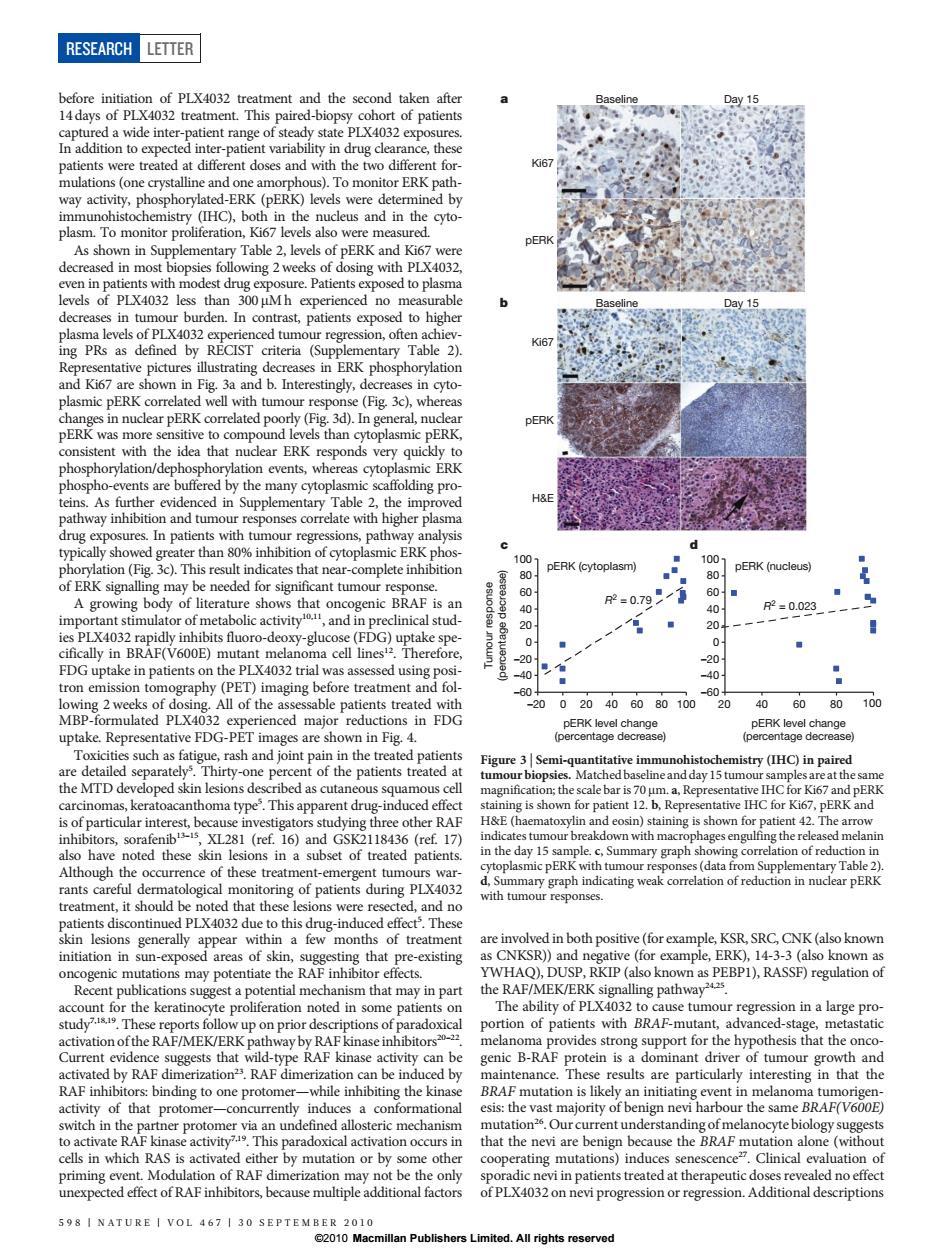
RESEARCH LETTER nent and the second taken afte 4032 treatm paired-b of patients er-pa rug cle ce t nulati To monitor erkr IHC oth in the ERE me decn ased in mos osing with PLX403 evels of PLX4032 es than 300 Mh able den. ghe PRs 6 ntary r DERK E y than ation e nts,where R Table 2.th H& In n er than 80 of cytopla pERK (cytoplasm) pERK (nucleus) e needed for sigr nt tumour 0.71 and in R=0.023 p-deaYgtcoscf uptake spe FDG upta in patients on the PLX4032 -20 All of the as 5 ted 20020406080100 20 406080 100 are sh aricitis suchas fai d tive in try(HC)in pa the MTD de ribed as cutaneous s day 15 tu th is 70 ative IHC for Ki6 PER H&E5 sshown for 17 e day 15 nary graph of rec (data e of these ent-emergent tun PERK wit油 d n LX4032 are involved in both nositive (forexample ksR SRC CNK (also know initiation in su that pre isting chanism that may RAF/MEK/ERN alling pathway tion of patients with BA-mutan activation of the RAF/MEK d by nce. results a inte esting in that the ational hat the nevi are by mutation or b oth ating mutatio indu nescenc Clinical 598 INATURE I VOL 467 1 30 ⊥.All rights reserved before initiation of PLX4032 treatment and the second taken after 14 days of PLX4032 treatment. This paired-biopsy cohort of patients captured a wide inter-patient range of steady state PLX4032 exposures. In addition to expected inter-patient variability in drug clearance, these patients were treated at different doses and with the two different formulations (one crystalline and one amorphous). To monitor ERK pathway activity, phosphorylated-ERK (pERK) levels were determined by immunohistochemistry (IHC), both in the nucleus and in the cytoplasm. To monitor proliferation, Ki67 levels also were measured. As shown in Supplementary Table 2, levels of pERK and Ki67 were decreased in most biopsies following 2 weeks of dosing with PLX4032, even in patients with modest drug exposure. Patients exposed to plasma levels of PLX4032 less than 300 mM h experienced no measurable decreases in tumour burden. In contrast, patients exposed to higher plasma levels of PLX4032 experienced tumour regression, often achieving PRs as defined by RECIST criteria (Supplementary Table 2). Representative pictures illustrating decreases in ERK phosphorylation and Ki67 are shown in Fig. 3a and b. Interestingly, decreases in cytoplasmic pERK correlated well with tumour response (Fig. 3c), whereas changes in nuclear pERK correlated poorly (Fig. 3d). In general, nuclear pERK was more sensitive to compound levels than cytoplasmic pERK, consistent with the idea that nuclear ERK responds very quickly to phosphorylation/dephosphorylation events, whereas cytoplasmic ERK phospho-events are buffered by the many cytoplasmic scaffolding proteins. As further evidenced in Supplementary Table 2, the improved pathway inhibition and tumour responses correlate with higher plasma drug exposures. In patients with tumour regressions, pathway analysis typically showed greater than 80% inhibition of cytoplasmic ERK phosphorylation (Fig. 3c). This result indicates that near-complete inhibition of ERK signalling may be needed for significant tumour response. A growing body of literature shows that oncogenic BRAF is an important stimulator of metabolic activity10,11, and in preclinical studies PLX4032 rapidly inhibits fluoro-deoxy-glucose (FDG) uptake specifically in BRAF(V600E) mutant melanoma cell lines12. Therefore, FDG uptake in patients on the PLX4032 trial was assessed using positron emission tomography (PET) imaging before treatment and following 2 weeks of dosing. All of the assessable patients treated with MBP-formulated PLX4032 experienced major reductions in FDG uptake. Representative FDG-PET images are shown in Fig. 4. Toxicities such as fatigue, rash and joint pain in the treated patients are detailed separately5 . Thirty-one percent of the patients treated at the MTD developed skin lesions described as cutaneous squamous cell carcinomas, keratoacanthoma type5 . This apparent drug-induced effect is of particular interest, because investigators studying three other RAF inhibitors, sorafenib13–15, XL281 (ref. 16) and GSK2118436 (ref. 17) also have noted these skin lesions in a subset of treated patients. Although the occurrence of these treatment-emergent tumours warrants careful dermatological monitoring of patients during PLX4032 treatment, it should be noted that these lesions were resected, and no patients discontinued PLX4032 due to this drug-induced effect5 . These skin lesions generally appear within a few months of treatment initiation in sun-exposed areas of skin, suggesting that pre-existing oncogenic mutations may potentiate the RAF inhibitor effects. Recent publications suggest a potential mechanism that may in part account for the keratinocyte proliferation noted in some patients on study7,18,19. These reports follow up on prior descriptions of paradoxical activation of the RAF/MEK/ERK pathway by RAF kinase inhibitors20–22. Current evidence suggests that wild-type RAF kinase activity can be activated by RAF dimerization23. RAF dimerization can be induced by RAF inhibitors: binding to one protomer—while inhibiting the kinase activity of that protomer—concurrently induces a conformational switch in the partner protomer via an undefined allosteric mechanism to activate RAF kinase activity7,19. This paradoxical activation occurs in cells in which RAS is activated either by mutation or by some other priming event. Modulation of RAF dimerization may not be the only unexpected effect of RAF inhibitors, because multiple additional factors are involved in both positive (for example, KSR, SRC, CNK (also known as CNKSR)) and negative (for example, ERK), 14-3-3 (also known as YWHAQ), DUSP, RKIP (also known as PEBP1), RASSF) regulation of the RAF/MEK/ERK signalling pathway24,25. The ability of PLX4032 to cause tumour regression in a large proportion of patients with BRAF-mutant, advanced-stage, metastatic melanoma provides strong support for the hypothesis that the oncogenic B-RAF protein is a dominant driver of tumour growth and maintenance. These results are particularly interesting in that the BRAF mutation is likely an initiating event in melanoma tumorigenesis: the vast majority of benign nevi harbour the same BRAF(V600E) mutation26. Our current understanding of melanocyte biology suggests that the nevi are benign because the BRAF mutation alone (without cooperating mutations) induces senescence27. Clinical evaluation of sporadic nevi in patients treated at therapeutic doses revealed no effect of PLX4032 on nevi progression or regression. Additional descriptions pERK level change (percentage decrease) Tumour response (percentage decrease) pERK level change (percentage decrease) R2 = 0.79 –60 –40 –20 0 20 40 60 80 100 –20 0 20 40 60 80 100 R2 = 0.023 –60 –40 –20 0 20 40 60 80 100 20 40 60 80 100 pERK (cytoplasm) pERK (nucleus) c d pERK Ki67 H&E Baseline Day 15 pERK Ki67 a Baseline Day 15 b Figure 3 | Semi-quantitative immunohistochemistry (IHC) in paired tumour biopsies. Matched baseline and day 15 tumour samples are at the same magnification; the scale bar is 70 mm. a, Representative IHC for Ki67 and pERK staining is shown for patient 12. b, Representative IHC for Ki67, pERK and H&E (haematoxylin and eosin) staining is shown for patient 42. The arrow indicates tumour breakdown with macrophages engulfing the released melanin in the day 15 sample. c, Summary graph showing correlation of reduction in cytoplasmic pERK with tumour responses (data from Supplementary Table 2). d, Summary graph indicating weak correlation of reduction in nuclear pERK with tumour responses. RESEARCH LETTER 598 | NATURE | VOL 467 | 30 SEPTEMBER 2010 ©2010 Macmillan Publishers Limited. All rights reserved