正在加载图片...
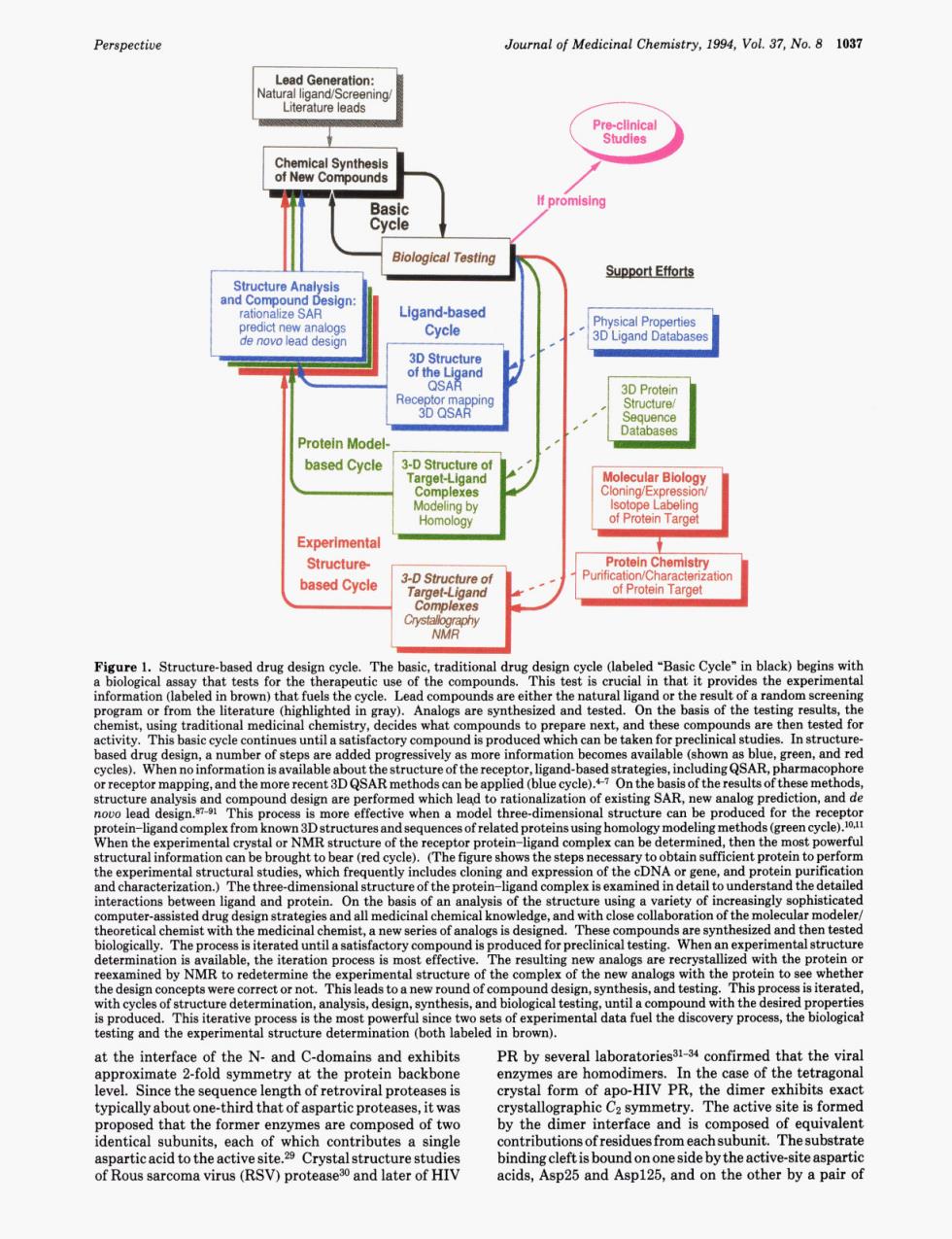
Perspective Journal of Medicinal Chemistry.1994.Vol 37.No.8 1037 Literature leads Biological Testing Support Efforts Re Protein Mode based Cycle MoliecularBlolog rotein Tame Experimenta rotein cle (labeled Cycle"in black)begins t pro the exper ie of th which inical 。14 are per the mpl hich The th pu clos on of the m d an the it tion p s is ectiv re the pr the eh orre to and tes cal tes ery p at the interfac f the N.and C-do and exhibits PR by s rol lahoratories31-34 ofirmed that the vira at the protein backbone are home HIV PR e of The activ e site is forme proposed that the fomere are como of tw by the mer interface aPatieacidtothea iteaspa7 )prote biadigdeniobondooneaideatheeig by a pa Perspective Journal of Medicinal Chemistry, 1994, Vol. 37, No. 8 1037 -port Efforts 3-0 struciure of Figure 1. Structure-based drug design cycle. The basic, traditional drug design cycle (labeled “Basic Cycle” in black) begins with a biological assay that tests for the therapeutic use of the compounds. This test is crucial in that it provides the experimental information (labeled in brown) that fuels the cycle. Lead compounds are either the natural ligand or the result of a random screening program or from the literature (highlighted in gray). Analogs are synthesized and tested. On the basis of the testing results, the chemist, using traditional medicinal chemistry, decides what compounds to prepare next, and these compounds are then tested for activity. This basic cycle continues until a satisfactory compound is produced which can be taken for preclinical studies. In structurebased drug design, a number of steps are added progressively as more information becomes available (shown as blue, green, and red cycles). When no information is available about the structure of the receptor, ligand-based strategies, including QSAR, pharmacophore or receptor mapping, and the more recent 3D QSAR methods can be applied (blue cycle).P7 On the basis of the results of these methods, structure analysis and compound design are performed which leqd to rationalization of existing SAR, new analog prediction, and de novo lead de~ign.~~-~l This process is more effective when a model three-dimensional structure can be produced for the receptor protein-ligand complex from known 3D structures and sequences of related proteins using homology modeling methods (green cycle).lOJ1 When the experimental crystal or NMR structure of the receptor protein-ligand complex can be determined, then the most powerful structural information can be brought to bear (red cycle). (The figure shows the steps necessary to obtain sufficient protein to perform the experimental structural studies, which frequently includes cloning and expression of the cDNA or gene, and protein purification and characterization.) The three-dimensional structure of the protein-ligand complex is examined in detail to understand the detailed interactions between ligand and protein. On the basis of an analysis of the structure using a variety of increasingly sophisticated computer-assisted drug design strategies and all medicinal chemical knowledge, and with close collaboration of the molecular modeler/ theoretical chemist with the medicinal chemist, a new series of analogs is designed. These compounds are synthesized and then tested biologically. The process is iterated until a satisfactory compound is produced for preclinical testing. When an experimental structure determination is available, the iteration process is most effective. The resulting new analogs are recrystallized with the protein or reexamined by NMR to redetermine the experimental structure of the complex of the new analogs with the protein to see whether the design concepts were correct or not. This leads to a new round of compound design, synthesis, and testing. This process is iterated, with cycles of structure determination, analysis, design, synthesis, and biological testing, until a compound with the desired properties is produced. This iterative process is the most powerful since two sets of experimental data fuel the discovery process, the biological testing and the experimental structure determination (both labeled in brown). at the interface of the N- and C-domains and exhibits approximate 2-fold symmetry at the protein backbone level. Since the sequence length of retroviral proteases is typically about one-third that of aspartic proteases, it was proposed that the former enzymes are composed of two identical subunits, each of which contributes a single aspartic acid to the active site.29 Crystal structure studies of Rous sarcoma virus (RSV) protease30 and later of HIV PR by several laboratories31-34 confirmed that the viral enzymes are homodimers. In the case of the tetragonal crystal form of apo-HIV PR, the dimer exhibits exact crystallographic Cz symmetry. The active site is formed by the dimer interface and is composed of equivalent contributions of residues from each subunit. The substrate binding cleft is bound on one side by the active-site aspartic acids, Asp25 and Asp125, and on the other by a pair of