正在加载图片...
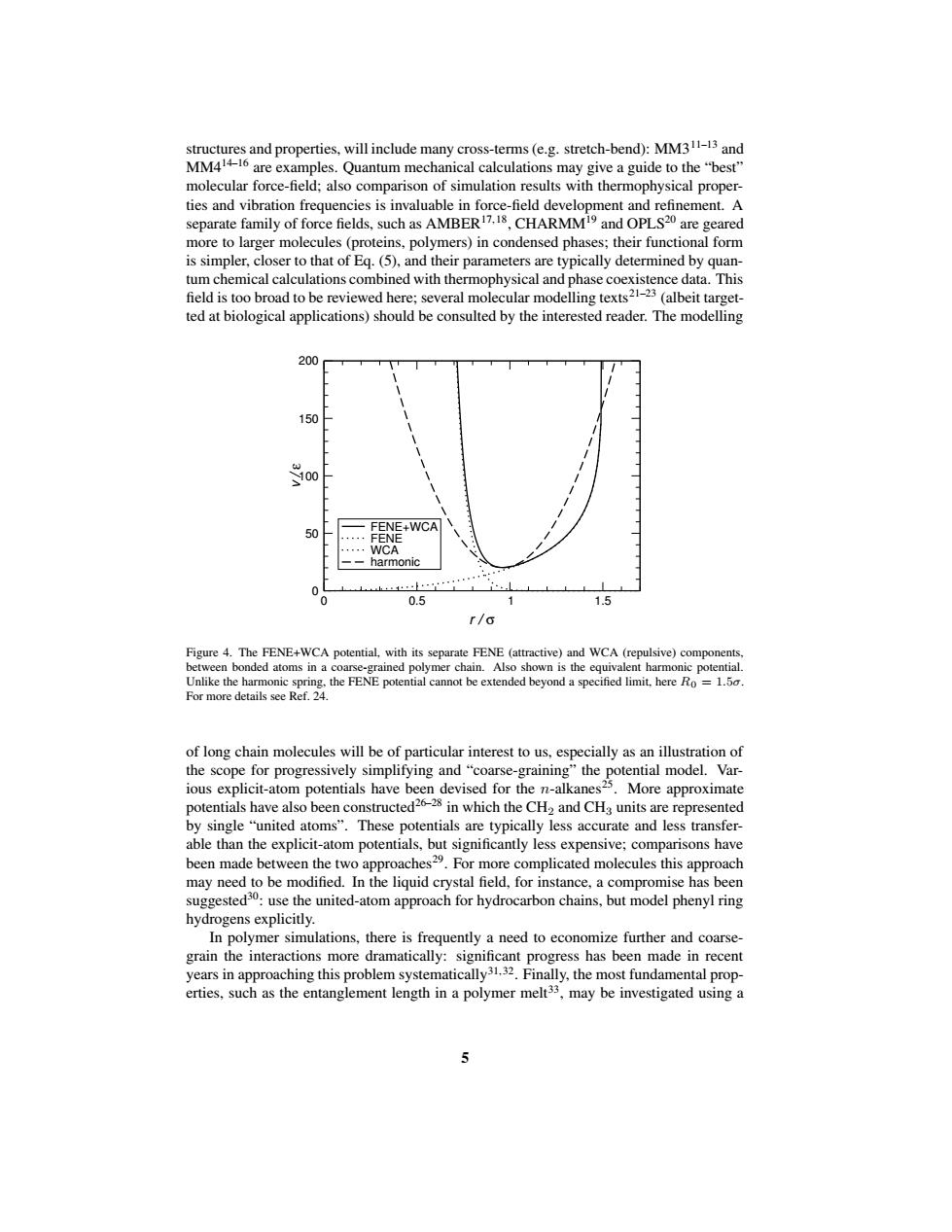
structures and properties,will include many cross-terms (e.g.stretch-bend):MM311-13 and MM414-16 are examples.Quantum mechanical calculations may give a guide to the"best" molecular force-field;also comparison of simulation results with thermophysical proper- ties and vibration frequencies is invaluable in force-field development and refinement.A separate family of force fields,such as AMBER7.18,CHARMM19 and OPLS20 are geared more to larger molecules(proteins,polymers)in condensed phases;their functional form is simpler,closer to that of Eq.(5),and their parameters are typically determined by quan- tum chemical calculations combined with thermophysical and phase coexistence data.This field is too broad to be reviewed here;several molecular modelling texts21-23(albeit target- ted at biological applications)should be consulted by the interested reader.The modelling 200 150 FENE+WCA 50 FENE ··WCA --harmonic 0 0.5 1.5 r/σ Figure 4.The FENE+WCA potential,with its separate FENE (attractive)and WCA (repulsive)components, between bonded atoms in a coarse-grained polymer chain.Also shown is the equivalent harmonic potential. Unlike the harmonic spring,the FENE potential cannot be extended beyond a specified limit,here Ro=1.5. For more details see Ref.24. of long chain molecules will be of particular interest to us,especially as an illustration of the scope for progressively simplifying and"coarse-graining"the potential model.Var- ious explicit-atom potentials have been devised for the n-alkanes25.More approximate potentials have also been constructed26-28 in which the CH2 and CHa units are represented by single "united atoms".These potentials are typically less accurate and less transfer- able than the explicit-atom potentials,but significantly less expensive;comparisons have been made between the two approaches29.For more complicated molecules this approach may need to be modified.In the liquid crystal field,for instance,a compromise has been suggested30:use the united-atom approach for hydrocarbon chains,but model phenyl ring hydrogens explicitly. In polymer simulations,there is frequently a need to economize further and coarse- grain the interactions more dramatically:significant progress has been made in recent years in approaching this problem systematically31.32.Finally,the most fundamental prop- erties,such as the entanglement length in a polymer melt33,may be investigated using a 5structures and properties, will include many cross-terms (e.g. stretch-bend): MM311–13 and MM414–16 are examples. Quantum mechanical calculations may give a guide to the “best” molecular force-field; also comparison of simulation results with thermophysical properties and vibration frequencies is invaluable in force-field development and refinement. A separate family of force fields, such as AMBER17, 18 , CHARMM19 and OPLS20 are geared more to larger molecules (proteins, polymers) in condensed phases; their functional form is simpler, closer to that of Eq. (5), and their parameters are typically determined by quantum chemical calculations combined with thermophysical and phase coexistence data. This field is too broad to be reviewed here; several molecular modelling texts21–23 (albeit targetted at biological applications) should be consulted by the interested reader. The modelling 0 0.5 1 1.5 r / σ 0 50 100 150 200 v / ε FENE+WCA FENE WCA harmonic Figure 4. The FENE+WCA potential, with its separate FENE (attractive) and WCA (repulsive) components, between bonded atoms in a coarse-grained polymer chain. Also shown is the equivalent harmonic potential. Unlike the harmonic spring, the FENE potential cannot be extended beyond a specified limit, here R0 = 1.5σ. For more details see Ref. 24. of long chain molecules will be of particular interest to us, especially as an illustration of the scope for progressively simplifying and “coarse-graining” the potential model. Various explicit-atom potentials have been devised for the n-alkanes25 . More approximate potentials have also been constructed26–28 in which the CH2 and CH3 units are represented by single “united atoms”. These potentials are typically less accurate and less transferable than the explicit-atom potentials, but significantly less expensive; comparisons have been made between the two approaches29 . For more complicated molecules this approach may need to be modified. In the liquid crystal field, for instance, a compromise has been suggested30: use the united-atom approach for hydrocarbon chains, but model phenyl ring hydrogens explicitly. In polymer simulations, there is frequently a need to economize further and coarsegrain the interactions more dramatically: significant progress has been made in recent years in approaching this problem systematically31, 32 . Finally, the most fundamental properties, such as the entanglement length in a polymer melt33 , may be investigated using a 5