正在加载图片...
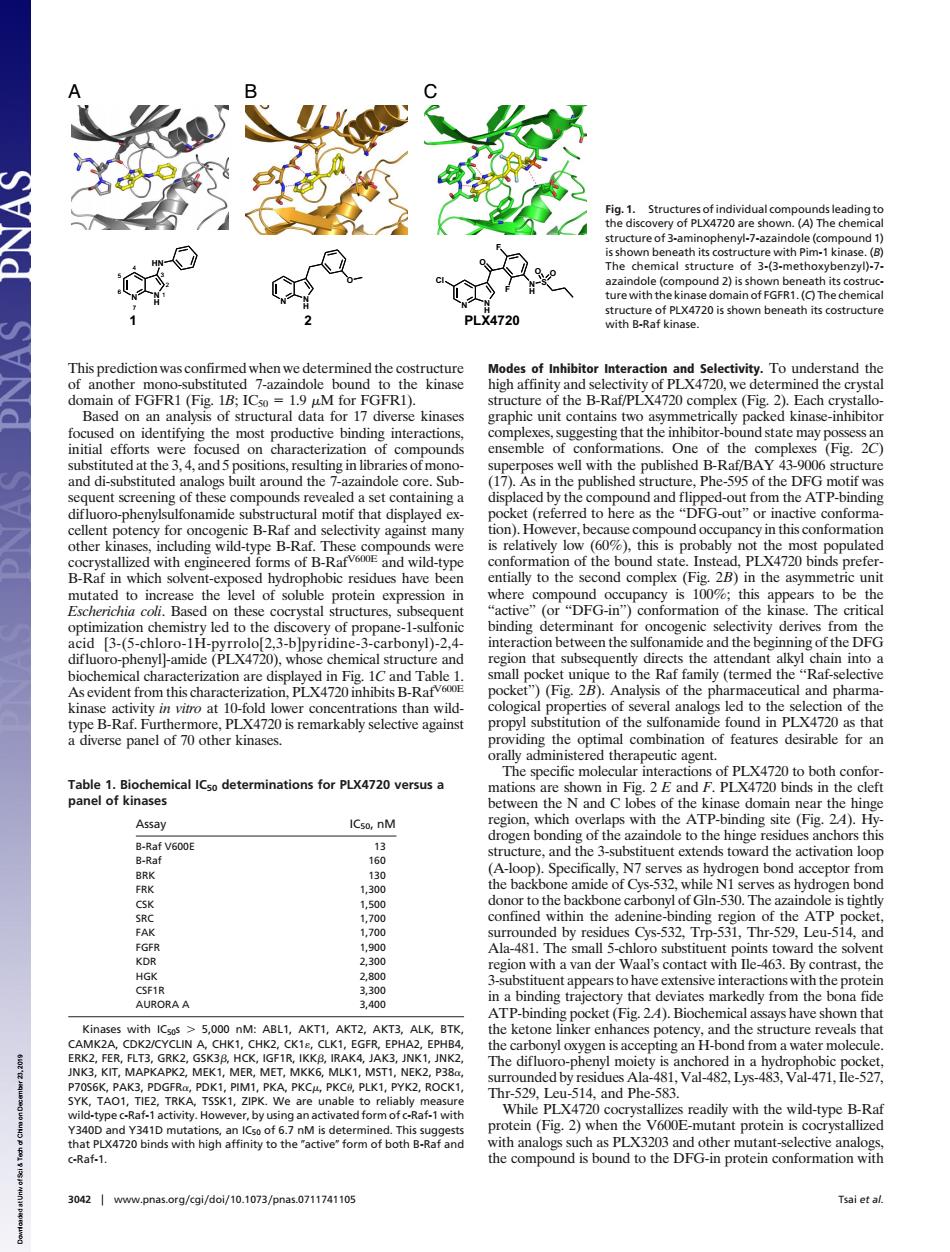
e (compo (C)The chemica PI X4720 B-ak of Inhib Each nding inte ng that the d5p ng in lil published B-Ra 17 dand fipped-out AT B-R B-Ra this co ype vith eng neered s of B Rarv.com ild-t n of th ted to i the evel of soluble protein ere ompound is to t n that sub B-Rav ket)(Fig.).Analysis of the ph 5y ind in PLX4720 a nical ICso determinations for PLX4720 versus a ations ATP-bindin Assa and ogen Gn-530. a-48 b r ●ys-5 T with aal's co h act with e. dly fre na fio with ICsos 5.00 M one ingd the The difluc at-1wit L and Phe both -an ein (Fig.2)when 720 nity to the c-Raf-1 30421www.pnas.oeg/kgi/do/10.1073/pnas.0711741105 Tsai et afThis prediction was confirmed when we determined the costructure of another mono-substituted 7-azaindole bound to the kinase domain of FGFR1 (Fig. 1B; IC50 1.9 M for FGFR1). Based on an analysis of structural data for 17 diverse kinases focused on identifying the most productive binding interactions, initial efforts were focused on characterization of compounds substituted at the 3, 4, and 5 positions, resulting in libraries of monoand di-substituted analogs built around the 7-azaindole core. Subsequent screening of these compounds revealed a set containing a difluoro-phenylsulfonamide substructural motif that displayed excellent potency for oncogenic B-Raf and selectivity against many other kinases, including wild-type B-Raf. These compounds were cocrystallized with engineered forms of B-RafV600E and wild-type B-Raf in which solvent-exposed hydrophobic residues have been mutated to increase the level of soluble protein expression in Escherichia coli. Based on these cocrystal structures, subsequent optimization chemistry led to the discovery of propane-1-sulfonic acid [3-(5-chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4- difluoro-phenyl]-amide (PLX4720), whose chemical structure and biochemical characterization are displayed in Fig. 1C and Table 1. As evident from this characterization, PLX4720 inhibits B-RafV600E kinase activity in vitro at 10-fold lower concentrations than wildtype B-Raf. Furthermore, PLX4720 is remarkably selective against a diverse panel of 70 other kinases. Modes of Inhibitor Interaction and Selectivity. To understand the high affinity and selectivity of PLX4720, we determined the crystal structure of the B-Raf/PLX4720 complex (Fig. 2). Each crystallographic unit contains two asymmetrically packed kinase-inhibitor complexes, suggesting that the inhibitor-bound state may possess an ensemble of conformations. One of the complexes (Fig. 2C) superposes well with the published B-Raf/BAY 43-9006 structure (17). As in the published structure, Phe-595 of the DFG motif was displaced by the compound and flipped-out from the ATP-binding pocket (referred to here as the ‘‘DFG-out’’ or inactive conformation). However, because compound occupancy in this conformation is relatively low (60%), this is probably not the most populated conformation of the bound state. Instead, PLX4720 binds preferentially to the second complex (Fig. 2B) in the asymmetric unit where compound occupancy is 100%; this appears to be the ‘‘active’’ (or ‘‘DFG-in’’) conformation of the kinase. The critical binding determinant for oncogenic selectivity derives from the interaction between the sulfonamide and the beginning of the DFG region that subsequently directs the attendant alkyl chain into a small pocket unique to the Raf family (termed the ‘‘Raf-selective pocket’’) (Fig. 2B). Analysis of the pharmaceutical and pharmacological properties of several analogs led to the selection of the propyl substitution of the sulfonamide found in PLX4720 as that providing the optimal combination of features desirable for an orally administered therapeutic agent. The specific molecular interactions of PLX4720 to both conformations are shown in Fig. 2 E and F. PLX4720 binds in the cleft between the N and C lobes of the kinase domain near the hinge region, which overlaps with the ATP-binding site (Fig. 2A). Hydrogen bonding of the azaindole to the hinge residues anchors this structure, and the 3-substituent extends toward the activation loop (A-loop). Specifically, N7 serves as hydrogen bond acceptor from the backbone amide of Cys-532, while N1 serves as hydrogen bond donor to the backbone carbonyl of Gln-530. The azaindole is tightly confined within the adenine-binding region of the ATP pocket, surrounded by residues Cys-532, Trp-531, Thr-529, Leu-514, and Ala-481. The small 5-chloro substituent points toward the solvent region with a van der Waal’s contact with Ile-463. By contrast, the 3-substituent appears to have extensive interactions with the protein in a binding trajectory that deviates markedly from the bona fide ATP-binding pocket (Fig. 2A). Biochemical assays have shown that the ketone linker enhances potency, and the structure reveals that the carbonyl oxygen is accepting an H-bond from a water molecule. The difluoro-phenyl moiety is anchored in a hydrophobic pocket, surrounded by residues Ala-481, Val-482, Lys-483, Val-471, Ile-527, Thr-529, Leu-514, and Phe-583. While PLX4720 cocrystallizes readily with the wild-type B-Raf protein (Fig. 2) when the V600E-mutant protein is cocrystallized with analogs such as PLX3203 and other mutant-selective analogs, the compound is bound to the DFG-in protein conformation with A C B N N H N H Cl S O F F O O N N H HN 1 2 3 4 5 6 7 N N H O 1 2 PLX4720 Fig. 1. Structures of individual compounds leading to the discovery of PLX4720 are shown. (A) The chemical structure of 3-aminophenyl-7-azaindole (compound 1) is shown beneath its costructure with Pim-1 kinase. (B) The chemical structure of 3-(3-methoxybenzyl)-7- azaindole (compound 2) is shown beneath its costructure with the kinase domain of FGFR1. (C) The chemical structure of PLX4720 is shown beneath its costructure with B-Raf kinase. Table 1. Biochemical IC50 determinations for PLX4720 versus a panel of kinases Assay IC50, nM B-Raf V600E 13 B-Raf 160 BRK 130 FRK 1,300 CSK 1,500 SRC 1,700 FAK 1,700 FGFR 1,900 KDR 2,300 HGK 2,800 CSF1R 3,300 AURORA A 3,400 Kinases with IC50s 5,000 nM: ABL1, AKT1, AKT2, AKT3, ALK, BTK, CAMK2A, CDK2/CYCLIN A, CHK1, CHK2, CK1, CLK1, EGFR, EPHA2, EPHB4, ERK2, FER, FLT3, GRK2, GSK3, HCK, IGF1R, IKK, IRAK4, JAK3, JNK1, JNK2, JNK3, KIT, MAPKAPK2, MEK1, MER, MET, MKK6, MLK1, MST1, NEK2, P38, P70S6K, PAK3, PDGFR, PDK1, PIM1, PKA, PKC, PKC, PLK1, PYK2, ROCK1, SYK, TAO1, TIE2, TRKA, TSSK1, ZIPK. We are unable to reliably measure wild-type c-Raf-1 activity. However, by using an activated form of c-Raf-1 with Y340D and Y341D mutations, an IC50 of 6.7 nM is determined. This suggests that PLX4720 binds with high affinity to the active form of both B-Raf and c-Raf-1. 3042 www.pnas.orgcgidoi10.1073pnas.0711741105 Tsai et al. Downloaded at Univ of Sci & Tech of China on December 23, 2019 ��������