正在加载图片...
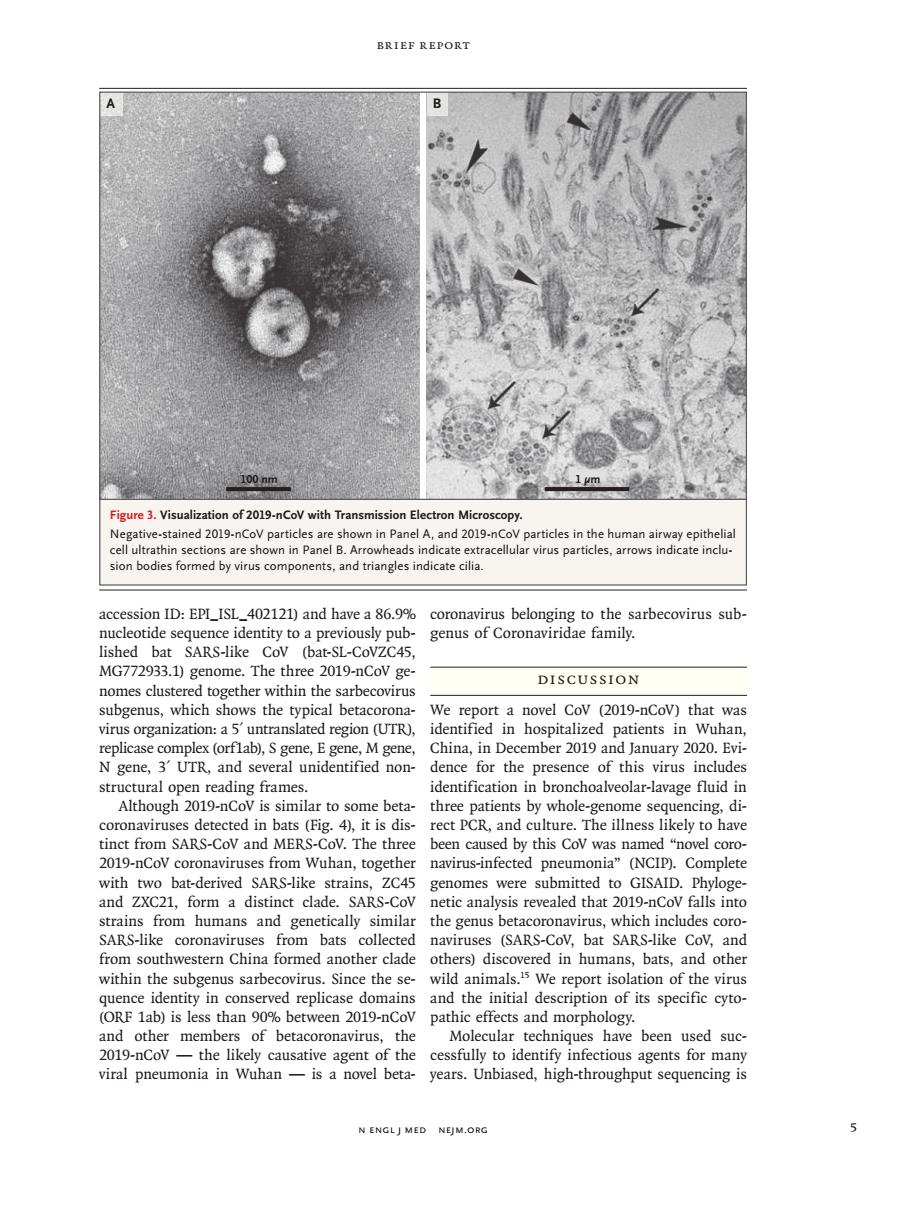
BRIEF REPORT Figure 3.Visualization of 2019-nCoy with Transmission Electron Mic sion bodies formed by virus components,and triangles indicate cilia acc on ID:EPI_ISL_402121)and havea86.9% MG772933.1)genome.The three 2019-nCoV ge DISCUSSION nomes clustered together within the sarbecovirus subgenus,which shows the typical betacorona- We report a novel Cov (2019-nCov)that was rins.oeganizatioaSlntnshcdtegonTRl identified in hospitalized patients in Wuhan, gene, three whole- age fluid coronavi ses detected in bats (Fig.4),it is dis-rect PCR.and culture.The illness likely to have tinct from SARS-CoV and MERS-CoV.The three been caused by this Cov was named "novel coro- 2019-nCov coronaviruses from Wuhan,together navirus-infected pneumonia"(NCIP).Complete with two bat-derived SARS-like strains,ZC45 genomes were submitted to GISAlD.Phyloge and ZXC21 form a distinct cla SA netic analysis revealed that 20 mans and gene from southwestern China formed another clade others)discovered in humans,bats,and other within the subgenus sarhecovirus since the se wild animals '5 we report isolation of the virus quence identity in conserved replicase domains and the initial description of its specific cyto- (ORF 1ab)is les than 90%between 2019-nCov pathic effects and morphology. of betacoronavirus, cular tec niques have been used suc pneumonia el beta cessfully ed,high -th N ENGLJ MED NEJM.ORG n engl j med nejm.org 5 Brief Report accession ID: EPI_ISL_402121) and have a 86.9% nucleotide sequence identity to a previously published bat SARS-like CoV (bat-SL-CoVZC45, MG772933.1) genome. The three 2019-nCoV genomes clustered together within the sarbecovirus subgenus, which shows the typical betacoronavirus organization: a 5′ untranslated region (UTR), replicase complex (orf1ab), S gene, E gene, M gene, N gene, 3′ UTR, and several unidentified nonstructural open reading frames. Although 2019-nCoV is similar to some betacoronaviruses detected in bats (Fig. 4), it is distinct from SARS-CoV and MERS-CoV. The three 2019-nCoV coronaviruses from Wuhan, together with two bat-derived SARS-like strains, ZC45 and ZXC21, form a distinct clade. SARS-CoV strains from humans and genetically similar SARS-like coronaviruses from bats collected from southwestern China formed another clade within the subgenus sarbecovirus. Since the sequence identity in conserved replicase domains (ORF 1ab) is less than 90% between 2019-nCoV and other members of betacoronavirus, the 2019-nCoV — the likely causative agent of the viral pneumonia in Wuhan — is a novel betacoronavirus belonging to the sarbecovirus subgenus of Coronaviridae family. Discussion We report a novel CoV (2019-nCoV) that was identified in hospitalized patients in Wuhan, China, in December 2019 and January 2020. Evidence for the presence of this virus includes identification in bronchoalveolar-lavage fluid in three patients by whole-genome sequencing, direct PCR, and culture. The illness likely to have been caused by this CoV was named “novel coronavirus-infected pneumonia” (NCIP). Complete genomes were submitted to GISAID. Phylogenetic analysis revealed that 2019-nCoV falls into the genus betacoronavirus, which includes coronaviruses (SARS-CoV, bat SARS-like CoV, and others) discovered in humans, bats, and other wild animals.15 We report isolation of the virus and the initial description of its specific cytopathic effects and morphology. Molecular techniques have been used successfully to identify infectious agents for many years. Unbiased, high-throughput sequencing is Figure 3. Visualization of 2019-nCoV with Transmission Electron Microscopy. Negative-stained 2019-nCoV particles are shown in Panel A, and 2019-nCoV particles in the human airway epithelial cell ultrathin sections are shown in Panel B. Arrowheads indicate extracellular virus particles, arrows indicate inclusion bodies formed by virus components, and triangles indicate cilia. A 100 nm 1 µm B